
Pédiatrie – Pneumo – HGE
Fiche réalisée selon le plan MGS
Item ECNi 43
Déf : la mucoviscidose est une maladie génétique de transmission autosomique récessive liée à une mutation du gène CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), résultant en une exocrinopathie généralisée.
Physiopathologie : le gène CFTR, situé sur le bras long du K7, code pour une protéine transmembranaire du même nom, intervenant dans la régulation du transport des ions chlorure Cl-.
La dysfonction ou l’absence de la protéine CFTR entraîne un défaut de transport du Cl- et une réabsorption hydro-sodée au niveau organes exocrines (arbre respiratoire, tube digestif et annexes, glandes sudoripares et tractus génital) qui produisent alors un mucus visqueux responsable des différents signes de la maladie.
Epidémiologie : maladie génétique autosomique récessive létale la plus fréquente en population caucasienne.
Incidence = 1 / 4700 naissances, d’où une prévalence de mutation CFTR hétérozygote estimée à 1 / 34 en population générale 1A. Sex-ratio = 1 mais expression plus marquée chez la femme 0
Génotypes : > 2000 mutations sont répertoriées, elles sont identiques sur les 2 allèles (homozygotie) ou différentes (hétérozygotie composite). En France, la F508del représente 70 % des cas. 1A
Les mutations sont classées selon leur statut pathogène 2
– « CF » associées à la mucoviscidose (dont F508del)
– « CFTR-RD » associées à la pathologie CFTR = effet modéré ou mineur
– « CF / CFTR-RD » associées à un large spectre phénotypique
– « Neutres = sans effet pathogène
– De signification clinique inconnue (nbses mutations faux-sens ou d’épissage)
Elles peuvent également être classées selon les anomalies de fonction CFTR 0
– Classe 1 : altération de la production de protéine, souvent non-sens ou délétion / insertion (absence partielle ou totale)
– Classe 2 : altération de la maturation cellulaire (dont F508del)
– Classe 3 : altération de la régulation, souvent dans les domaines de liaison à l’ATP
– Classe 4 : altération de la conduction du canal, domaines transmembranaires
– Classe 5 : altération de la stabilité de l’ARNm CFTR
– Classe 6 : altération de la stabilité de la protéine mature
Schématiquement, les 3 premières classes sont des mutations « sévères » (correspondant souvent à des mutations CF), les classes 4 et 5 sont « peu sévères » (correspondant souvent à des mutations CFTR-RD).
| Clinique | Paraclinique |
|---|---|
| Exocrinopathie généralisée : BPCO et autres complications respiratoires, insuffisance pancréatique | Dépistage néonatal systématique, DPN sur point d’appel Confirmation : test à la sueur + analyse CFTR |
A ) Clinique
Terrain / FdR
– 25 % de mucoviscidose dans la progéniture d’un couple à 2 parents hétérozygotes
– Pas de mutation de novo (3 cas rapportés) 0
Principaux signes cliniques chez l’enfant
– Signes respiratoires et infectieux persistants (75 % dès la 1ère année de vie) : toux, bronchorrhée, bronchiolites, bronchites « asthmatiformes »
– Signes digestifs : stéatorrhée et cassure de la courbe staturo-pondérale (insuffisance pancréatique exocrine 95%), iléus méconial, ictère cholestatique du nouveau-né
– Autres : cf. complications
B ) Paraclinique
-
Diagnostic positif
Test à la sueur +++ : examen de confirmation diagnostique de référence, mesure la concentration en Cl- de la sueur
– N < 30 mmol/L
– Mucoviscidose : valeur ≥ 60 mmol/L confirmée par une 2e mesure
– Valeurs intermédiaires (30-59 mmol/L) : répéter le test ± autres examens de cette section
Génétique (complément nécessaire du test de la sueur, mais ce n’est pas l’outil de confirmation 1A) : détermine le génotype du patient par
– Recherche des mutations les plus fréquentes
– ± Etude plus extensive (séquençage du gène complet)
Mesures électrophysiologiques (en centre spécialisé) : différence de potentiel nasal (DPP), courant de court-circuit (CCC) sur biopsie rectale.
-
Diagnostic prénatal (DPN) ciblé
Le diagnostic est spécifiquement recherché lors d’une grossesse dans 2 cas
– Signes échographiques évocateurs – en l’absence d’histoire familiale
– Situations de conseil génétique – ATCD du couple ou familial (cf. partie 4 Prévention)
Les signes échographiques évocateurs de mucoviscidose sont
– Hyperéchogénicité intestinale
– Dilatation digestive
– Non-visualisation de la vésicule biliaire
– Images évoquant une péritonite méconiale
L’étude génétique des 2 parents est le plus souvent pratiquée à ce stade, mais l’étude génétique préalable du fœtus est une option selon le contexte obstétrical (l’absence de mutation fréquente chez le fœtus alors considéré comme une non-atteinte).
Le DPN est une étape nécessairement invasive (donc à risque de perte foetale) réalisée par
– Choriocentèse (si mutations parentales identifiées +++)
– Amniocentèse au terme de 18 SA (si 1er enfant atteint et mutations parentales non-identifiées, de plus en plus rare) : dosage des immunoenyzmes intestinales, PAL et γGT, leucine aminopeptidase
– Diagnostic pré-implantatoire dans les situations de PMA 2
-
Dépistage néonatal systématique
Le dépistage systématique à J3 par dosage de la TIR (trypsine immunoréactive), est intégré aux tests au buvard néonataux ; cette enzyme est un marqueur de souffrance pancréatique peu spécifique (20 % de faux +) mais assez sensible (3 % de faux -) 1A
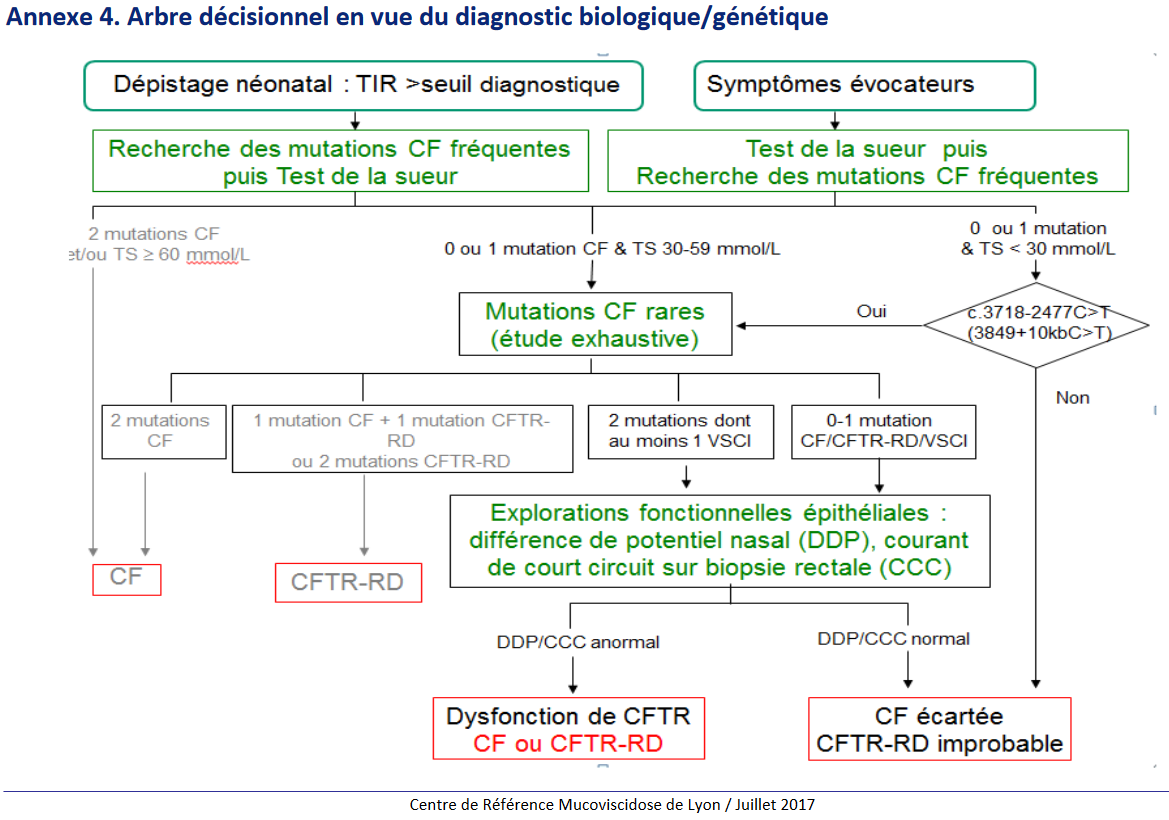
Les sources utilisées pour cette fiche sont équivoques dans la démarche diagnostique. Dans tous les cas, le test à la sueur est réalisé après la recherche de mutations fréquentes dans le cas du dépistage néonatal.
CAT devant un dosage TIR à J3 > 65 µg / L : analyse CFTR
– Absence de mutation fréquente : répéter le dosage TIR à J21 (si le TIR J3 était > 100 µg/L), sinon fin de procédure de dépistage 1A
– Si 1 ou 2 mutations CFTR, ou si le dosage TIR à J21 est supérieur au seuil (40 µg/L) : test à la sueur 1A. Le test à la sueur semble systématique pour tout dosage TIR supérieur au seuil diagnostique. 2
– S’il existe 0 ou 1 mutation CF associée à un test de la sueur mesuré à 30-59 mmol/L ; ou s’il existe une mutation c.3718-2477C>T* avec un test de la sueur négatif < 30 mmol/L : recherche de mutations rares. 2
* Note 0 : cette mutation semble être un cas particulier important. Elle est citée dans le texte du document HAS comme une indication au test génétique dans la fratrie asymptomatique d’un patient atteint de mucoviscidose, même en présence d’un test de la sueur négatif.
C ) Synthèse diagnostique 2
A) Histoire naturelle
La morbidité et la mortalité sont essentiellement d’origine respiratoire et infectieuse (90%) : l’évolution se fait vers une BPCO chez le grand enfant. La colonisation bactérienne des sécrétions bronchiques survient dès les premiers jours de vie avec des germes banals, mais la colonisation à P. Aeruginosa marque un tournant évolutif majeur dans l’histoire naturelle de la muciviscidose.
La médiane de survie d’un enfant né en 2010 avec la mucoviscidose est d’environ 50 ans, en nette progression.
B) Complications
Colonisation bactérienne chronique des sécrétions bronchiques (signe cardinal)
– Persistance à l’ECBC ou aux prélèvements respi. profonds d’une colonisation > 6 mois (culture + sur ≥ 3 prélèvements espacés chacun d’au moins 1 mois) sans signes d’infection
– D’abord H. Influenzae et S. Aureus
– P. aeruginosa = tournant évolutif
Autres complications respiratoires
– Signes de BCPO : sd obstructif irréversible aux EFR d’aggravation progressive
– Distension thoracique et emphysème plus tardifs avec sd restrictif aux EFR
– Dilatation des bronches, hémoptysies, pneumothorax
– Insuffisance respiratoire chronique : hypoxie nocturne puis diurne, dyspnée d’effort puis permanente, décompensation tardive avec hypercapnie et hypertension pulmonaire
– Infections mycobactériennes (M. abscessus, M. avium), aspergillaires (aspergillose broncho-pulmonaire allergique)
Insuffisance pancréatique exocrine +++
– Carences en vitamines liposolubles (ADEK) et oligoéléments
– Retard staturo-pondéral, dénutrition chez le grand enfant
– Déminéralisation osseuse (hypovitaminose D = rachitisme) 2
– Pancréatite chronique / épisodes de pancréatite aiguë 2
Autres complications digestives
– Atteinte hépatobiliaire : stéatose, lithiases multiples, cirrhose biliaire multifocale (5-15%) évoluant vers l’HTP et plus rarement l’IHC
– RGO
– Prolapsus rectal
– Invagination intestinale iléo-colique 2
– Complication des ATB 2 : colite à C. difficile, syndrome de dysbiose
– Mucocèle appendiculaire (spécifique) 2 : douleur en FID + image échographique
– Syndrome d’obstruction intestinale distale (spécifique) 2 : douleur aiguë en FID, troubles du transit allant de la constipation à un tableau occlusif
Complications génitales
– Infécondité masculine constante par atrésie des canaux déférents
– Hypofertilité féminine
– Retard pubertaires
Complications endocriniennes et métaboliques
– Intolérance au sucre, diabète insulinodépendant (par évolution de la pancréatite chronique 0)
– Déshydratation aiguë par hyponatrémie typiquement sur coups de chaleur
Complications ORL : sinusite maxillaire, polypose nasale
Complication cardiaque : cardiomyopathie non obstructive
Complications urologiques ² : incontinence urinaire, lithiases rénales, insuffisance rénale
Complications psychiatriques ² : anxiété, syndrome dépressif, troubles phobiques ou obsessionnels…
A ) Bilan
-
Bilan initial 2
Cette évaluation recherche des complications, elle est adaptée à l’âge du patient, aux circonstances diagnostiques et aux signes cliniques. Un bilan complet est généralement réalisé au 6e mois.
| Bilan devant une mucoviscidose |
|---|
| Evaluation respiratoire : clinique, ECBC, Rx thoracique, EFR 0 Evaluation nutritionnelle (dès la fin du 2e mois, répétée au moins à 1 et 2 ans) Fonction pancréatique exocrine : clinique, élastase fécale Selon les signes d’appel et l’âge : exploration fonctionnelle d’exercice annuelle à partir de 10 ans, investigations ORL, intestinales ou hépatobiliaires, pancréatique endocrine… |
-
Conseil génétique 1A
Il permet la reconstitution de l’arbre généalogique, le dépistage parental dans le but d’évaluer les risques futurs, et d’envisager les stratégies de prévention (cf. Terrain / FdR, et fiche Diagnostic pré-natal et IMG).
Principales indications
– Couples avec ATCD d’enfant atteint de mucoviscidose (2 parents forcément hétéroZ)
– Situation d’hétérozygotie connue de l’un des parents (dépistage du 2e parent)
– Couples ayant des apparentés proches avec enfant atteint (dépistage des 2 parents)
A l’issue du test génétique
– Les couples n’ayant aucune mutation fréquente doivent être rassurés
– Les couples avec 2 parents hétérozygotes se voient proposer un DPN
– En cas de mucoviscidose évoquée sur signes échographiques, et de mutation fréquente retrouvée chez 1 seul parent, un séquençage complet du gène chez l’autre parent recherche une mutation rare (20 % dans cette situation).
L’obligation d’information à la parentèle s’applique dans ce cadre 2 : la personne peut refuser de transmettre elle-même les informations à ses proches à risque, mais elle est alors tenue de communiquer les coordonnées des intéressés au médecin.
B ) Traitement
Suivi et PEC multidisciplinaire dans des centres de ressources et de compétences pour la mucoviscidose (CRCM). La PEC au domicile est favorisée pour un maximum de situations (par exemple, mise à disposition de sets de perfusion d’antibiotiques).
PEC médicale
| Domaine de PEC | Mesures utiles |
|---|---|
| Respiratoire et infectieuse |
Kiné respiratoire systématique, souvent précédée d’aérosols mucolytiques comme la rhDNase (Pulmozyme®) ou de sérum salé hypertonique ATBthérapie adaptée aux germes de l’ECBC pour 2-3 semaines (selon germe, gravité clinique ou lésionnelle, niveau de résistance… Traitement systématique des primo-infections et infections chroniques à P. Aeruginosa 2 Au stade d’insuffisance respiratoire chronique : OLD, VNI, transplantation pulmonaire |
| Digestive |
Insuffisance pancréatique exocrine : opothérapie (extraits pancréatiques gastroprotégés) Atteinte hépatobiliaire : acide urodésoxycholique |
| Nutritionnelle |
Régime hypercalorique (apports énergétiques journaliers = 120-150 % des reco habituelles) Supplémentation : vitamines ADEK systématique, Na+ selon déperditions Avant diversification alim. : supplémenter le lait avec 2 mEq/kg de Na+ |
Note 1A : les thérapies protéiques (Ivacaftor, Lumacaftor® 2), visant à agir sur l’origine moléculaire de la maladie, ouvrent des perspectives très encourageantes et pourraient améliorer de beaucoup la survie.
PEC sociale et paramédicale
– ALD 100 %
– Soutien psychologique
– Education thérapeutique (cf. prévention) ± associations (Vaincre la mucoviscidose), projet d’accueil individualisé…
C) Prévention
Mesures générales
– Hygiène domestique, éviction du tabac, réduction de la pression allergénique
– Mode de garde individuel plutôt que collectif la 1ère année de vie
– Activité physique adaptée aux performances respiratoires
Vaccinations ciblées
– Grippe annuelle
– Pneumocoque 23-valent après 2 ans
– Hépatite A
– ± Varicelle après 1 an
Suivi au CRCM 2
– Tous les mois jusqu’à 6 mois, tous les 2 mois jusqu’à 1 an
– Puis au moins 4 consultations / an pour les formes typiques de l’enfant ou de l’adulte
– Bilan paraclinique détaillé annuel (mais ECBC et spirométrie à chaque consultation)



