
Pédiatrie
Fiche réalisée selon le plan MGS
Item ECNi x
Déf : la syndrome de Rett est une maladie neuro-développementale d’origine génétique, caractérisée dans sa forme typique par un décélération du développement psycho-moteur survenant après une phase de développement normal.
Epidémiologie
– Maladie quasi-exclusivement féminine, 2e cause de déficience intellectuelle chez la fille après la T21
– Prévalence de 1 / 10.000 à 1 / 22.000 naissances féminines
Génétique : la plupart des formes atypiques n’ont pas de cause moléculaire identifiée
– Mutation MECP2 (localisé en Xq28) : 95 % des formes typiques, certaines formes atypiques (avec préservation du langage ++)
– Mutation CDKL5 : formes atypiques avec épilepsie précoce
– Mutation FOXG1 : formes atypiques congénitales
| Clinique | Paraclinique |
|---|---|
| Période de régression suivie d’une phase de récupération ou stabilisation (dans tous les cas) Rett typique : les 4 critères majeurs et aucun critère d’exclusion Rett atypique : ≥ 2 critères majeurs et ≥ 5 critères accessoires ; 3 variants cliniques |
Rett typique : analyse ciblée MECP2 ou séquençage panel / exome Rett atypique : analyse MECP2 si préservation du langage, CDKL5 si épilepsie précoce, FOXG1 si forme congénitale |
A ) Clinique
-
Critères diagnostiques
Le syndrome de Rett est un diagnostic clinique reposant sur l’association chez une même patiente de caractéristiques cliniques, d’une séquence évolutive particulière et de l’exclusion de certains diagnostics différentiels. Les critères complets sont disponibles en annexe de la source 2.
Le diagnostic de syndrome de Rett nécessite
– Une période de régression suivie d’une phase de récupération ou stabilisation (dans tous les cas)
– Les 4 critères majeurs et aucun critère d’exclusion = Rett classique
– ≥ 2 critères majeurs et ≥ 5 critères accessoires = Rett atypique.
| Critères majeurs | Critères accessoires | Critères d’exclusion |
|---|---|---|
| 1) Perte partielle ou complète de l’utilisation volontaire des mains. 2) Perte partielle ou complète du langage oral 3) Troubles de la marche : instabilité ou absence de marche 4) Stéréotypies manuelles caractéristiques (pression, torsion, applaudissement, mains dans la bouche, frottement, lavage des mains) |
1) Dysfonctionnement respiratoire avec des épisodes d’apnées pendant la veille, de blockpnée et d’expulsion forcée d’air 2) Bruxisme pendant la veille 3) Troubles du sommeil 4) Troubles du tonus 5) Troubles vasomoteurs 6) Scoliose / cyphose 7) Retard de croissance 8) Mains et pieds petits, hypotrophiques 9) Rires et pleurs immotivés 10) Diminution de la sensibilité à la douleur 11) Communication par le regard – pointage visuel |
1) Lésions cérébrales d’origine périnatale ou postnatale, troubles métaboliques ou maladie neurologique progressive 2) Développement psychomoteur anormal pendant les 6 premiers mois de la vie. |
-
Formes typiques ou classiques (75%)
Séquence évolutive typique avant 4 ans
– Perte d’acquis psycho-moteurs entre 1 et 2 ans (utilisation des mains, babillage, communication dont parfois interaction visuelle) durant quelques semaines / mois
– Puis période de « réveil » : récupération relative des capacités de communication visuelle sans récupération motrice franche
– On peut observer des stéréotypies manuelles, des traits autistiques avec perte de contact puis apparition vers 3 ans d’un pointage oculaire, avec décélération de la croissance du PC
Eléments d’apparition plus tardive
– Après 4-6 ans : pointage oculaire contrastant avec l’absence de langage, troubles ventilatoires, retard de croissance avec mains et pieds hypertrophiques, cyphose, scoliose
– Chez l’adolescente / l’adulte : petite taille, rigidité parkinsonienne avec troubles trophiques (altération précoce peau et ongles), dégradation de l’humeur notamment après 25 ans
-
Formes atypiques ou variantes (25%)
Il existe, en plus des critères diagnostiques de syndrome de Rett atypique, des signes cliniques de 3 variants définis, orientant par la suite la recherche d’altération génétique.
Signes de Rett atypique avec épilepsie précoce (Hanefeld variant) : chez une petite fille (et plus rarement le garçon)
– Epilepsie précoce se manifestant par des orages de crises cloniques ou toniques avant 5 mois
– Evolution avant 1 an vers des spasmes infantiles (syndrome de West)
– Epilepsie myoclonique pharmacorésistante
– Retard du développement sévère
– Stéréotypies manuelles
Signes de Rett atypique congénital (Rolando variant) : chez la fille ou le garçon
– Retard du développement sévère
– Microcéphalie congénitale ou post natale précoce à partir de 4 mois
– Mouvements anormaux à type de dyskinésie, dystonie mobile généralisée
– Dyskinésies bucco-linguales
Signes de Rett atypique avec préservation du langage (Zappela variant) : chez la fille
– Parfois dans le bilan de troubles autistiques : absence de langage avec persistance de mots ou phrases écholaliques, stéréotypies fréquentes, atteinte motrice mineure
– Les autres signes classiques de syndrome de Rett (microcéphalie, retard de croissance, épilepsie) sont absents
Note : d’autres formes appelées « syndromes de Rett-like » peuvent être diagnostiquées devant l’association d’un retard de développement (± régression psychomotrice), d’une décélération de croissance du PC, de stéréotypies manuelles et d’un pointage visuel.
B ) Paraclinique
Génétique moléculaire (confirmation)
Rett typiques
– Séquençage ciblé de MECP2 par Sanger avec recherche de grands réarrangements par MLPA par exemple
– Ou séquençage haut débit d’un panel de gènes ou de l’exome
Rett atypiques : pas de cause identifiée le plus souvent !
– Avec préservation du langage : analyse MECP2
– Avec épilepsie précoce : analyse CDKL5
– Forme congénitale : analyse FOXG1
C ) Diagnostic différentiel
Rett typique
– Syndrome d’Angelman : déficit intellectuel sévère, absence de langage, rires immotivés, stéréotypies avec battements des mains, microcéphalie, ataxie
– Syndrome de Pitt Hopkins : déficit intellectuel sévère, absence de langage, rires immotivés, troubles ventilatoires, microcéphalie, ataxie, particularités morphologiques
– Troubles du spectre autistique avec régression transitoire
Rett atypique congénital
– Encéphalopathie avec mouvements anormaux (chorée, athétose, dyskinésie, dystonie faciales ou orolinguales…)
– Encéphalopathie avec microcéphalie congénitale d’origine génétique (microcéphalies à gyrations simplifiées)
– Encéphalopathies épileptiques précoces (STXBP1 ++)
– Encéphalopathies par anomalie du métabolisme des neurotransmetteurs
Rett atypique avec épilepsie précoce : encéphalopathies épileptiques précoces génétiques
– Monogéniques (STXBP1 ++)
– Malformations cérébrales (lissencéphalie)
– Métaboliques (épilepsies sensibles aux vitamines B6, B9)
Il existe une grande variabilité clinique tout au long de la vie du patient, avec une évolution du phénotype de la petite enfance à l’âge adulte. Ces aspects évolutifs (non-repris ici) sont largement développés dans le PNDS.
A ) Bilan initial
-
Bilan initial
| Bilan initial d’un syndrome de Rett |
|---|
| Evaluation intellectuelle, psycho-motrice et de la communication – Motricité globale : Rett Syndrome Gross Motor Scale – Utilisation des mains : score composite d’utilisation des mains – Echelles d’évaluation plus générales : score sévérité clinique du syndrome de Rett, échelle d’impression globale clinique, échelle d’évaluation Motrice-Comportementale |
| Recherche de comorbidités – signes associés – Neurologiques : épilepsie (EEG, vidéo-EEG), évaluation des stéréotypies et mouvements anormaux – Evaluation de la commande respiratoire – Recherche de troubles du sommeil – Recherche d’un allongement du QT (ECG) – Recherche de troubles du comportement – Recherche de troubles de l’alimentation et du transit (± FOGD, TODG, radio-cinéma) – Evaluation de la croissance (objectif IMC autour du 25e percentile) – Recherche d’une ostéoporose (ostéodensitométrie) – Recherche d’une anomalie musculo-squelettique : scoliose, subluxation de hanche – Recherche d’anomalies dentaires |
| Recherche de contre-indications médicamenteuses : CI habituelles si QT long, utilisation de dépresseurs respiratoires prudente (BZD, morphiniques et dérivés) |
-
Conseil génétique
L’analyse génétique des 2 parents, si elle est négative, est en faveur d’une mutation de novo et donc d’un risque de récidive très faible : environ 1 % pour les parents d’un enfant Rett avec mutation dans l’un des 3 gènes associés – MECP2, CDKL5 et FOXG1 (mosaïcisme germinal).
Pour MECP2 et CDKL5, la recherche de mutation peut être effectuée chez des sœurs asymptomatiques d’un enfant atteint, car une inactivation biaisée du chromosome X porteur de l’allèle muté peut exceptionnellement expliquer des cas de porteuses asymptomatiques.
En cas de nouvelle grossesse, on peut proposer un diagnostic prénatal aux couples qui le souhaitent.
B ) Traitement
Il n’existe aucun traitement spécifique du syndrome de Rett. PEC multidisciplinaire !
> PEC du retard de développement / polyhandicap
– Kinésithérapie active et passive
– Psychomotricité
– Orthophonie, éducation à la communication non-orale
– Autres : musicothérapie, balnéothérapie, équithérapie, ostéopathie, recours au psychologue
> Appareillage : en lien avec l’orthopédiste et l’orthoprothésiste
> Troubles du comportement : évaluation psychologique ± psychiatrique, TCC, techniques de relaxation, psychotropes en cas d’agitation majeure
> Troubles du sommeil : mélatonine et neuroleptiques sont souvent nécessaires, appareillage d’un syndrome d’apnées du sommeil
> Epilepsie : pas de recommandation spécifique. Le syndrome de Rett est une bonne indication au régime cétocène, préférentiellement chez les patients porteurs d’une gastrostomie.
> Dénutrition : régime hypercalorique si retard de croissance, discuter la gastrostomie dans les formes sévères (IMC < 3e percentile) malgré le régime hypercalorique
> Fragilité osseuse : supplémentation en calcium et vitamine D, lutte contre l’immobilité, PEC d’une ostéoporose
> RGO : IPP ± anti-H2 voire béthanéchol (si résistance à la bithérapie en l’absence d’épilepsie ou asthme instable), parfois chirurgie anti-reflux
> Complications respiratoires
– Positionnement de la tête, patchs de scopolamine ± injection de toxine des glandes salivaires,
– Si pneumopathies récidivantes : ATBprophylaxie alternée (amox, augmentin, bactrim, macrolides)
– Kiné respiratoire ± aides techniques (Cough assist, Percussionnaire) voire VNI / O2thérapie nocturne
C ) Suivi
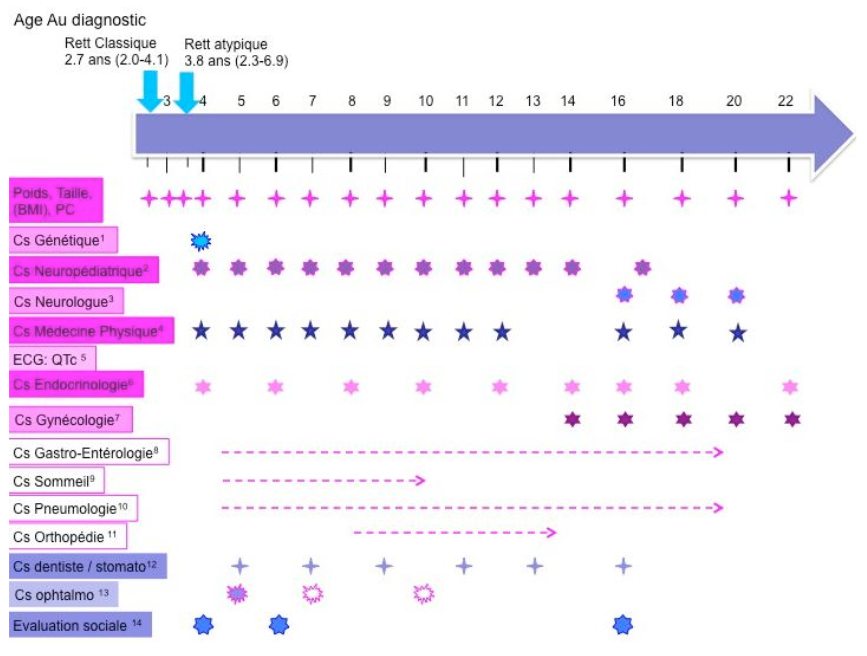
Un bilan complémentaire est en plus indiqué de façon annuelle
– Systématique : NFS, ferritine, albumine, pré-albumine, protides, créatininémie, ionogramme et bilan phosphocalcique, calciurie, créatininurie 25-OH-vitamineD3, PAL
– Si IMC < 25e percentile ou prise d’anti-épileptiques : vitamines B9 et B12
– Si régime cétocène : bilan lipidique, bilan hépatique et pancréatique
– Si ≥ 50 % des apports caloriques par voie entérale (sonde entérale, gastrostomie) : compléter le BH, magnésémie
– Si aggravation des crises épileptiques sous traitement / suspicion d’effets secondaires : dosage des anti-épileptiques



